
Company
Platform
Insights

In the fight against cancer, early treatments were often blunt and non-specific, relying on broad-spectrum chemotherapy, radiation, and invasive surgeries that targeted not just tumors but also healthy tissues. These generalized approaches, while sometimes effective, left many patients with few options when their specific cancer didn't respond.
As our understanding of cancer has advanced, we've moved from broad classifications like "lung cancer" to more precise definitions based on cell types (e.g., adenocarcinoma) and genetic mutations (e.g., KRAS mutations). This shift has enabled more targeted therapies, such as EGFR inhibitors for specific lung cancers and HER2-targeted treatments for certain breast cancers.
A game changer in cancer treatment has been the introduction of immunotherapies, starting with the FDA approval of the first antitumor cytokine called interferon-alpha 2 (IFN-a2) in 1986. They work by “employing” the body's own immune system to fight tumors, offering new options where traditional therapies fall short.
Modern immunotherapies include immune checkpoint inhibitors like pembrolizumab (Keytruda) and nivolumab (Opdivo), which block the PD-1/PD-L1 pathway. This blockade enables T cells, the organism's most powerful natural “warheads”, to attack cancer cells more effectively.
Other approaches, such as CAR-T cell therapy, genetically engineer a patient’s own T cells to target specific cancer antigens, achieving remarkable results in blood cancers like leukemia and lymphoma.
While being powerful in some cases, immunotherapies unfortunately work in only about 12.5% of cancer patients. The key question here, then, is how to know if someone is in that % or not. Another good question is how to increase the overall % of responders by creating better immunotherapies.
Complex diseases like cancer are rarely driven by a single gene or protein; instead, they are influenced by a myriad of genetic, cellular, and tissue-level processes that vary across different patient populations. This variability makes it difficult to predict how a drug will affect an individual patient, particularly in cases like immunotherapies, where the pathways are poorly understood.
At Noetik, we believe our best chance to improve cancer care is to model the entirety of biological systems to generate a picture of the complexity of cancer progression. Only then can we answer the key question of drug discovery: which drugs, for which patients.
To make a real impact on drug discovery, we need computational capabilities that can understand and simulate disease biology at the patient level, identifying the right targets and matching them with the right therapies.
In our previous technical report we introduced OCTO, our Artificial Intelligence engine for simulating patient biology. At its core, OCTO is a transformer-based model similar to the Large Language and Vision Models that power ChatGPT but designed for biological data. In the same way those models learn to understand text and generate images by training on user generated content on the web, we designed OCTO to understand tumor biology by training on data generated from cancer patients in our lab.
Because biology lacks a single abstraction, like language, OCTO is designed to integrate data across modalities - including spatial omics, DNA sequencing, multiplex protein staining and histological staining - to learn a unified representation of tumor biology, that can be queried at the patient level.
These unique capabilities are made possible by new experimental techniques and a new kind of data foundation that we have built from the ground up in Noetik's labs.
Noetik has developed a cutting-edge data factory specifically to create multimodal biological datasets, purpose-built for self-supervised learning, that offer an unprecedented biological depth to our models. Starting with non-small cell lung cancer (NSCLC), we have completed our first spatial multi omics atlas of human tumor biology—a comprehensive mapping of over 1,000 lung cancer cases from the level of the tissue to the genome.
This data resource is built on the power of paired multimodal spatial omics, combining these data with clinically relevant techniques like hematoxylin and eosin (H&E) and whole exome sequencing (WES) to capture a remarkably broad spectrum of cancer biology—all from the same patient samples.
Our process begins with high-quality human tumor specimens, meticulously preserved and prepared, and sourced directly to our in-house biobank.
To generate our spatial multi omics data data, we first use a custom protein panel to map the complex interactions within the tumor-immune microenvironment (TME) and gather structural data to train self-supervised models.
Next, the same tissue slide undergoes H&E staining, offering the model a detailed view akin to that of a pathologist.
Just four micrometers away, we analyze adjacent tissue sections using spatial transcriptomics to capture RNA expression at the single-cell level.
Finally, we perform whole exome sequencing on a sample from each tumor to identify key genetic alterations.
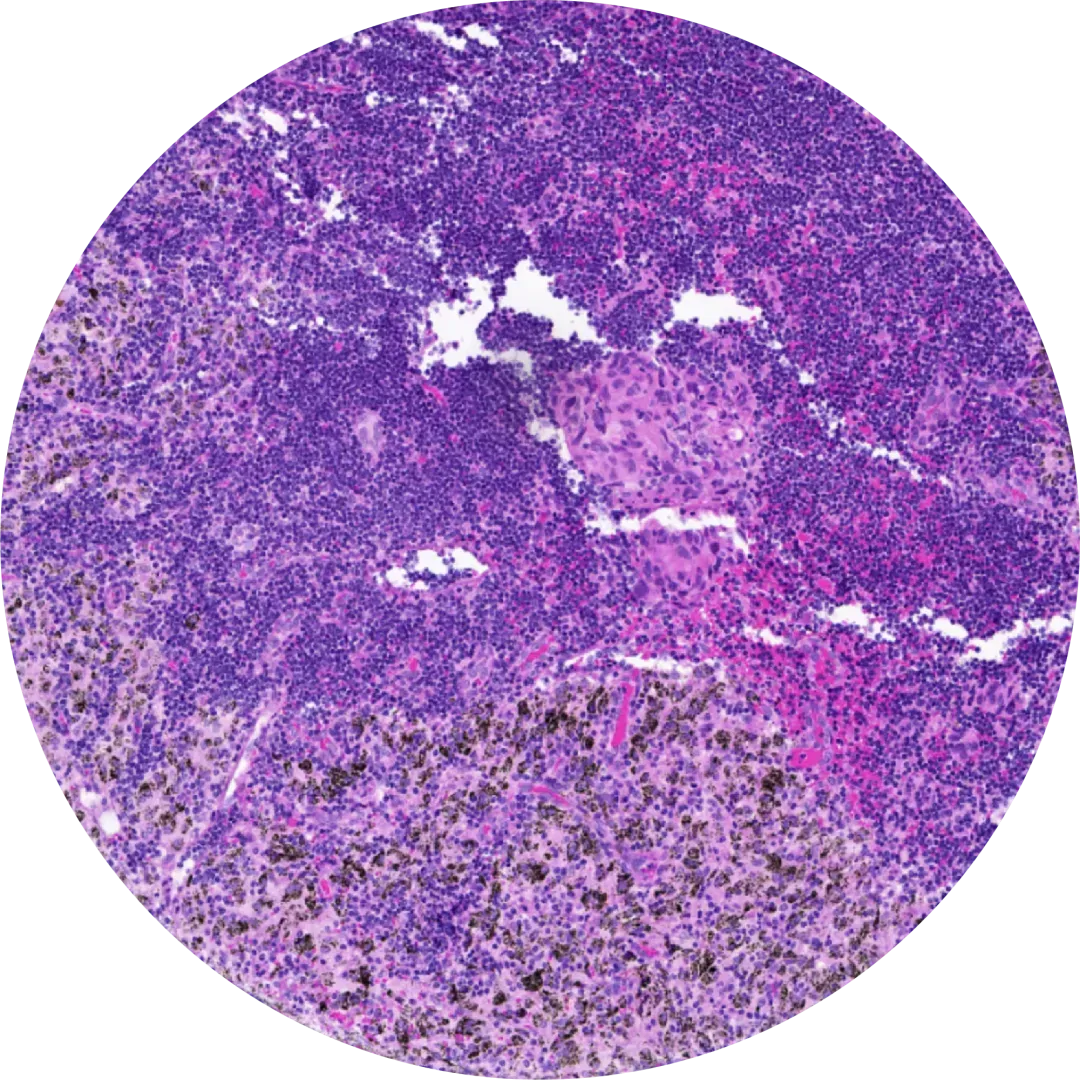
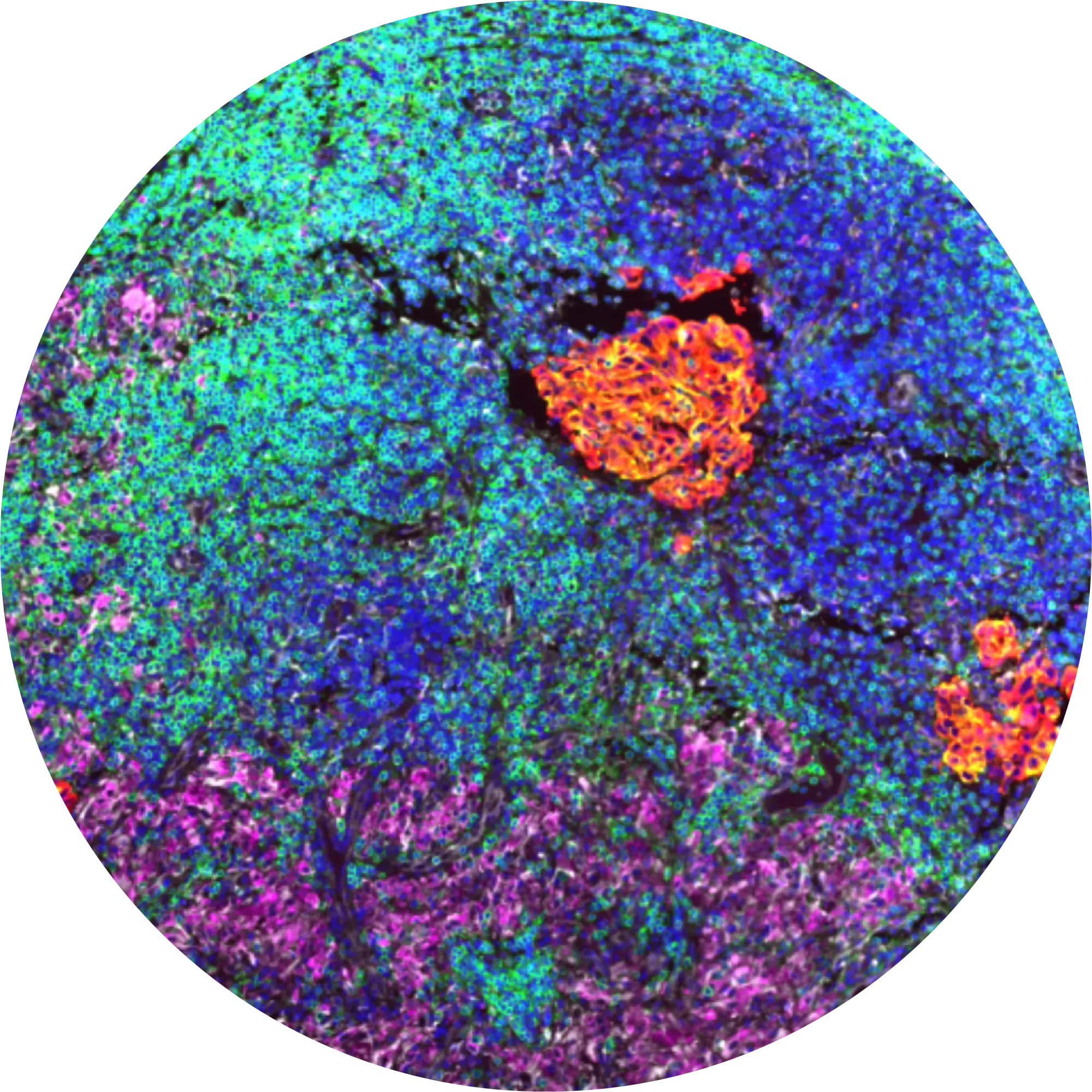
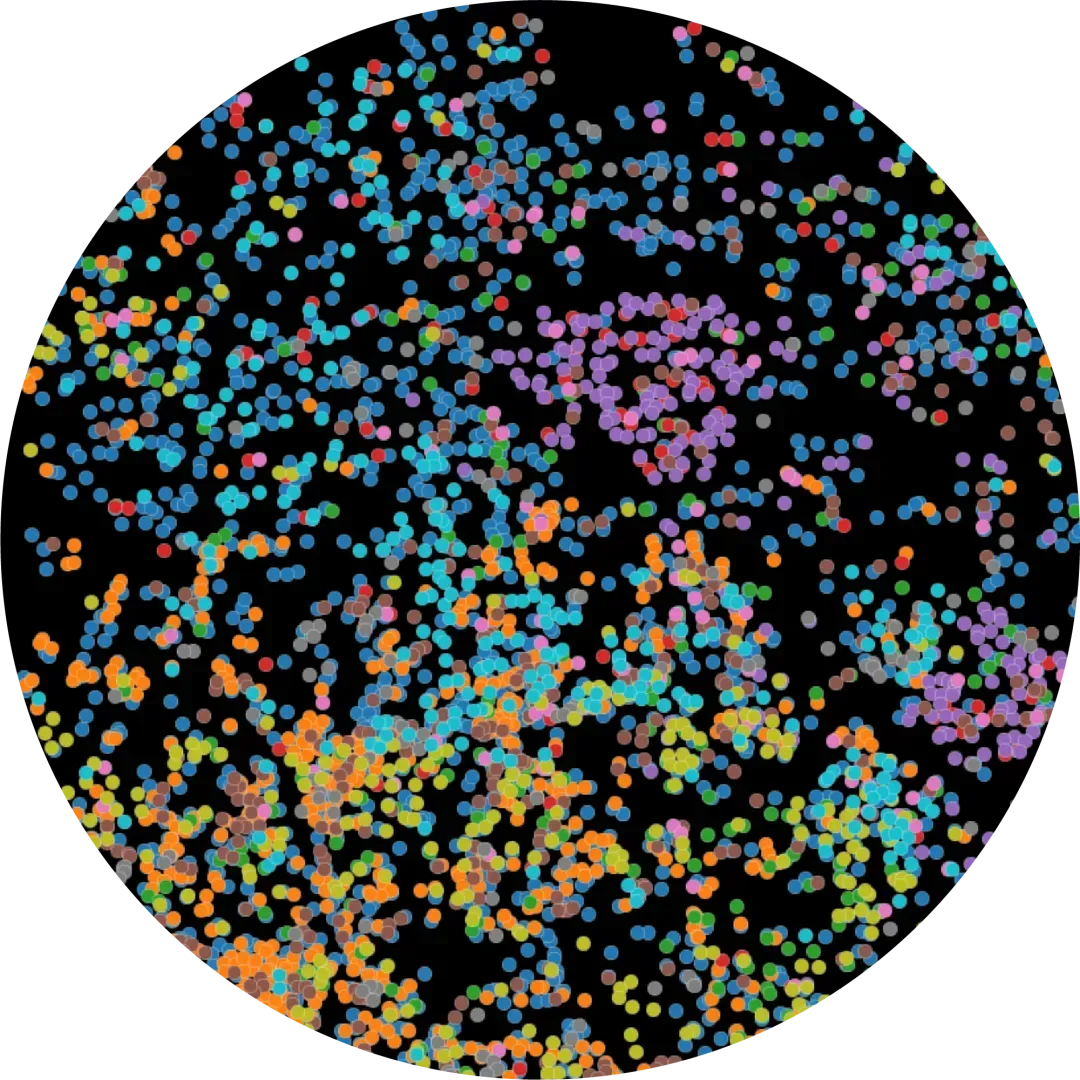
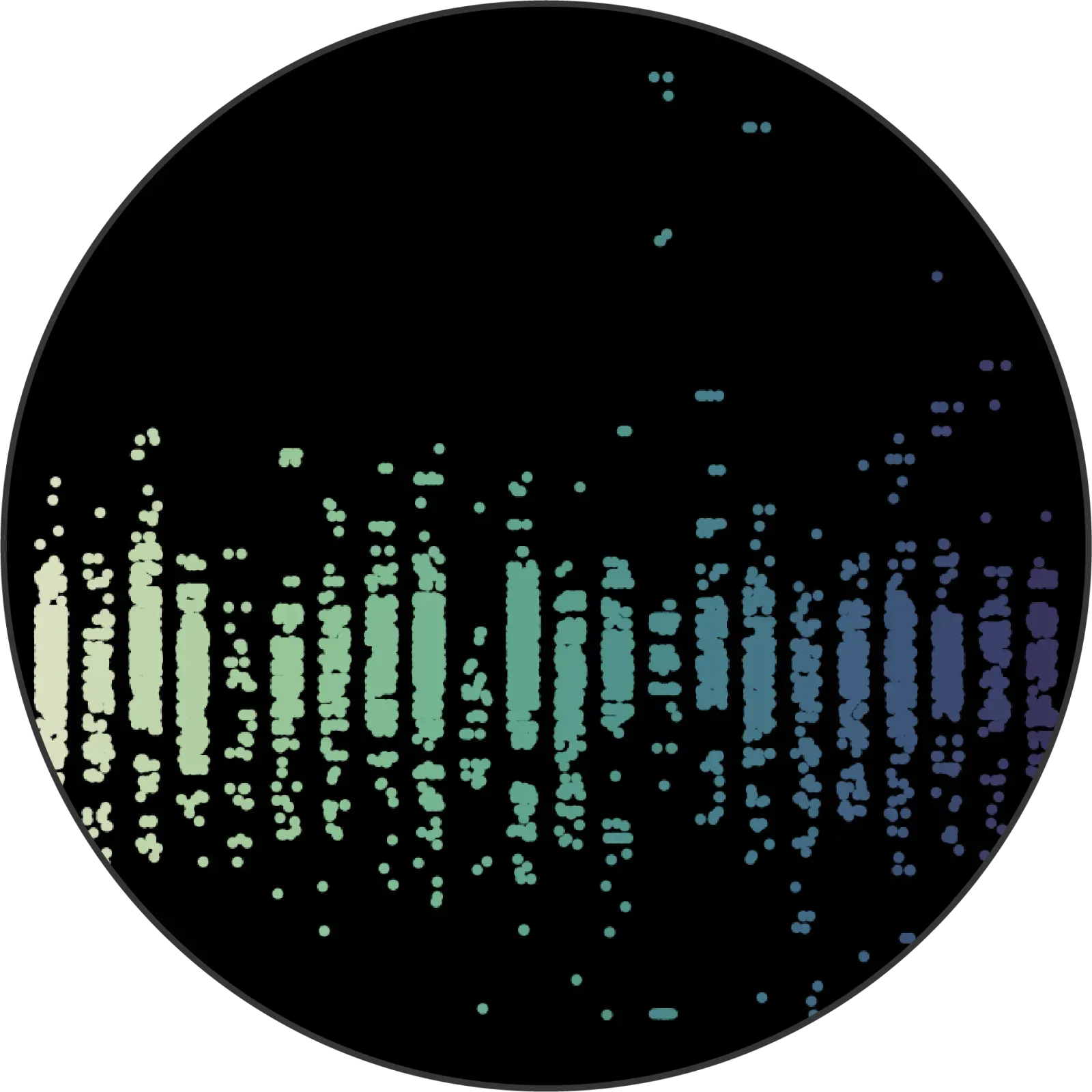
This entire process is purpose-built to optimize data for machine learning.
Traditional digital pathology often falls short because models can easily pick up on slide-level artifacts, especially when only a single slide represents each patient.
To overcome this, we’ve designed our dataset so that each patient is represented by dozens of individual samples, spatially randomized alongside control tissues, spread across different slides and staining batches, with consistent quality control at every step. We’ve developed our own patent pending processes to construct these tissue microarrays (TMA). This ensures that our models focus on true biological signals, not on irrelevant variations introduced during slide preparation.
One of our strategic edges comes from how we generate and handle data at Noetik, our data quality protocols.
We take the extra step of sourcing entire tumor samples ourselves for our internal biobank, allowing us to carefully control every aspect of the sample quality and data generation process. We’re selective, using only tissues that meet our stringent standards for things like sample age, ischemia time, and percent necrosis, among other parameters. Before any sample becomes part of our multimodal atlas, it’s meticulously reviewed by a board-certified pathologist.

Noetik’s lung cancer data is more than just a collection of data points. It’s a carefully curated, global snapshot of NSCLC, with a balanced representation of male and female patients and tumors sourced from six continents to reflect the true diversity of human disease.
The dataset is a product of the first data factory designed for self-supervised learning from first principles. This means that the data itself is the training label. Instead of trying to make an inferential leap all the way from high dimensional biological data to drug response, or focusing on a specific aspect of patient biology, Noetik trains transformers on the complex biological relationships inherent to cells and tissues. By analogy, rather than training on dissociated bags of words, we learn language from books.
The challenges of developing drugs reflect the complexity of human biology. To grapple with this complexity we need new tools, and new methods - we need to turn the complexity of biological systems on its head, and use it as the substrate for training a new class of models. This worldview is reflected in the intentional construction of all of our data- which is uniquely controlled and multimodal to fuel the development of this next class of models, and ultimately a next generation of drugs catering to more specific disease biology.
What truly sets our data apart is its direct connection to clinical outcomes. Around 10% of the dataset is built from tumor samples paired with long-term follow-up data, including patient responses to treatments like immune checkpoint inhibitors.
This clinical linkage is crucial—it allows us to train self-supervised models not just to analyze tumor phenotypes, but to predict how real patients will respond to specific therapies. By integrating these outcomes, we’re able to develop predictive biomarkers and uncover mechanisms of therapeutic resistance, ultimately bridging the gap between data and patient care. We are able to develop models that solve the most crucial clinical question: which patients will benefit from which drug.
The powerful self-supervised models, trained on our one-of-a-kind spatial multi omics data, opens access to a wealth of applications from drug discovery, to translational science, even clinical diagnostics and digital pathology.
Our data factory and our self-supervised learning engine OCTO are specifically designed and validated to be efficient in uncovering new therapeutic targets, identifying biomarkers, and helping to identify the right patients for our clinical trials and those of our partners.
But there’s more data here than we can tackle on our own. That’s why we’re looking to team up with leading academic organizations, companies, and industry partners to accelerate the progress in cancer research and bring impactful solutions to patients faster.
We’re rapidly branching out into areas across the spectrum of oncology, with early data already in areas such as sarcoma, ovarian cancer, and colorectal cancer. Plus, we’re layering in advanced data like in vivo CRISPR perturbation, starting with lung adenocarcinoma, through our Perturb-Map platform.
Ultimately our aim is to train the most comprehensive foundation models of cell and tissue biology to understand human disease, from a combination of deeply multimodal profiled human samples and scaled in vivo perturbations and pharmacology. Because we believe that the best cancer drugs, begin from the patient.










